Table of Contents
ICHとは
ICHとは、International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use(医薬品規制調和国際会議)の略称です。
ICHは、医薬品規制当局と製薬業界の代表者が協働して、医薬品規制に関するガイドラインを科学的・技術的な観点から作成する国際会議で、他に類がない場となっています。ICHは、1990年の創設以来、グローバル化する医薬品開発・規制・流通等に対応するべく、着実に進化を遂げてきました。ICHの使命は、限られた資源を有効に活用しつつ安全性・有効性及び品質の高い医薬品が確実に開発され上市されるよう、より広範な規制調和を世界的に目指すことです。
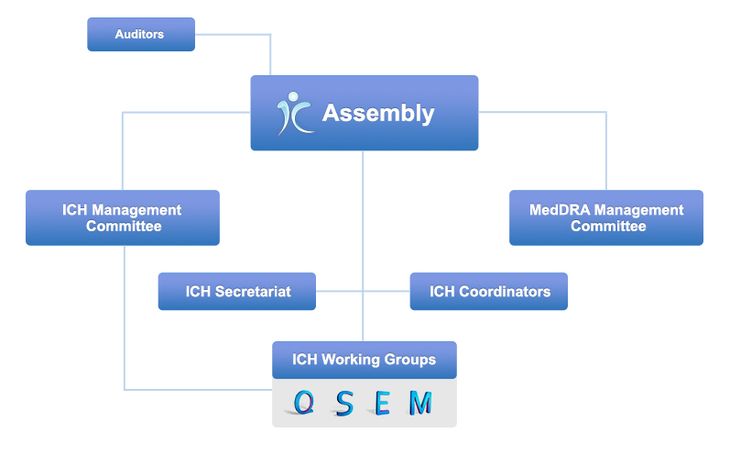
2015年10月23日、ICHはスイス法人化に伴い、組織再編をしました。その結果、現在のICHは、全ての参加メンバーで構成され法人の主体となる総会(Assembly)、総会での議論の準備や法人の運営を担う管理委員会(Management Committee)、専門家がガイドラインの議論を行う各作業部会(Working Group)等から成り立っています。
ICHの概要、設立、および目的
ICHとは、医薬品規制調和国際会議の略称であり、正式名称は International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use です。これは、世界各国の医薬品規制当局と製薬業界の代表者が集まり、薬事規制に関するガイドラインを作成する国際会議です。
ICHは1990年4月に、日本、米国、およびEU(創始三極)それぞれの医薬品行政当局と製薬産業界の代表の計6団体によって創設されました。その目的は、医薬品の承認審査や市販後安全対策などに関するガイドラインを作成し、薬事規制の国際調和を推進することです。特に、新薬の研究開発中に実施された試験を繰り返す必要性を低減または排除するため、技術指針と製品登録の要件の解釈と適用における一層の調和を目指しています。
国際調和によって、以下のような達成を目指しています:
- 不必要な臨床試験の重複を減らすこと。
- 資源を節約し、安全性・有効性・品質の高い新薬の開発・上市を促進すること。
- 患者が継続的に新しい治療を利用できるようにすること。
- 安全性・有効性のない、不必要な動物試験を減少させること。
- ヒト、ヒト以外の動物、および資源をより経済的に利用し、世界的な開発の不必要な遅れや新薬品の入手可能性を排除し、品質、安全性、効能、および公衆衛生を守ること。
ICHの最終的な目標は、グローバルな視点から、新医薬品をより早く患者に届け、公衆衛生に貢献することです。
2015年10月23日にはスイス法に基づき国際的な非営利法人となり、組織再編を行いました。
ICH発足の経緯
日本・米国・ヨーロッパでは、医薬品の販売開始前に政府による評価・承認を行うため、それぞれ独自に法制度を整備してきました。特に1960年代から1970年代にかけては、各国で急速に法令やガイドラインが整備され、新医薬品の品質、有効性および安全性についてのデータ報告・評価の体制が整いました。しかし、新医薬品の品質、有効性、安全性を評価するという基本は共通であったものの、承認申請の際の詳細な技術的要件は地域により異なっていました。製薬企業の国際化に伴い、各地域の規制要件を満たすため、時間とコストのかかる重複した試験を数多く行う必要がありました。
そのため、拡大する医薬品開発コストへの懸念を背景に、必要な患者に安全で有効な新医薬品をより早く提供するため、各地域の医薬品承認審査の基準の合理化・標準化が必要となり、1990年4月、日本・米国・ヨーロッパの各医薬品規制当局と業界団体の6者によりICHが発足しました。
ICHは発足以降、毎年2回会合を続けています。また、ICH会合後、各地域でICHの公開シンポジウムが開催されています。
新医薬品の品質・有効性・安全性の評価にかかわる技術的なガイドラインだけでなく、最近では承認申請資料の形式、市販後安全体制などの調和も進めており、またICHに参加していない国・地域との交流、情報の共有化が進んでいます。
ICHのメンバーと組織構成
ICHは主に以下のメンバー団体で構成されています:
- 創設規制当局メンバー: 厚生労働省/医薬品医療機器総合機構(MHLW/PMDA)、米国食品医薬品局(FDA)、欧州委員会/欧州医薬品庁(EC/EMA)。これらの創設規制当局メンバーは、主要事項に関して拒否権を持っています。
- 創設産業界メンバー: 日本医薬工業協会(JPMA)、米国研究製薬工業協会(PhRMA)、欧州製薬団体連合会(EFPIA)。
- 常任規制当局メンバー: ヘルスカナダ、スイスメディック。
- 規制当局メンバー: ブラジル国家衛生監督庁(ANVISA)、中国国家医薬品監督管理局(NMPA/CFDA)、シンガポール保健科学庁(HAS)、韓国食品医薬品安全処(MFDS)、台湾食品薬物管理署(TFDA)など。
- 産業界メンバー: バイオテクノロジーイノベーション協会(BIO)、国際ジェネリック・バイオシミラー医薬品協会(IGBA)、世界セルフケア連盟(GSCF/WSMI)など。
2019年11月時点で、16団体がメンバーとして参加しています。
決定権や議決権を持たないオブザーバーも参加しており、世界保健機関(WHO)、国際製薬団体連合会(IFPMA)など、2019年11月時点で32団体が参加しています。WHOとIFPMAは常任オブザーバーでもあります。
現在のICHの組織は、総会(Assembly)、管理委員会(Management Committee)、**専門家がガイドラインの議論を行う各作業部会(Working Group)**等から成り立っています。総会は全ての参加メンバーで構成され、法人の主体となります。管理委員会は総会での議論の準備や法人の運営を担います。MedDRA管理委員会も存在し、ICHの管理や政策決定、トピック選択、進捗監視などを行います。
ICHの成果物
ICHの主な成果物は、医薬品の品質、安全性、有効性、および複合領域に関するガイドラインです。新規ガイドラインの作成に加え、既存ガイドラインの改定や維持管理、ガイドラインを補完する補遺やQ&Aなども作成されています。
ステップ4で合意されたガイドラインは、各国が原則として「変更せずにそのまま取り込む」ことが求められます。これにより、各国行政当局は国内実施に向けた規制や法的な整備を行う必要があります。
ICH設立以来、多くのガイドラインが作成されており、例えば、新医薬品の承認申請資料の様式と内容を調和する「コモンテクニカルドキュメント」(CTD)は、2000年11月に最終合意に至った重要な成果物の一つです。
ガイドライン作成プロセス
ICHガイドラインは以下の5つのステップを経て作成されます。
- ステップ1: 提案されたトピックについて、運営委員会で承認を受けた後、作業部会が設置され、技術文書案が作成されます。
- ステップ2: 技術文書案が作業部会で合意に至り、運営委員会で承認されるとステップ2aに到達します。その後、行政側がガイドライン案として承認することでステップ2bに到達します。
- ステップ3: 各地域(日本、米国、EU)の規制当局からガイドライン案が公表され、意見募集が行われます。寄せられた意見を作業部会で検討し、ガイドライン案を修正します。
- ステップ4: ガイドライン案が運営委員会の規制当局代表者によって最終的に採択され、日本、米国、EUの3者による合意文書として完成します。
- ステップ5: 各国・地域において、国内の手続きに従ってガイドラインが実施されます。日本では厚生労働省から通知されます。
ICHに対するPMDAの関与
独立行政法人医薬品医療機器総合機構(PMDA)は、日本の厚生労働省(MHLW)と共に、日本の医薬品規制当局を代表する主要メンバーとしてICHに設立当初から深く関与し、その活動に積極的に貢献しています。
1. 意思決定への参加:
PMDAは、ICHの最高意思決定機関である総会(Assembly)や、ICHの運営方針や戦略的な議論を行う管理委員会(Management Committee)に代表者を派遣し、ICH全体の方向性決定や重要事項の議論に参加しています。これにより、日本の立場や意見をICHの運営に反映させています。
2. ガイドライン策定への貢献:
PMDAの各分野(品質、安全性、有効性、複合領域など)の専門職員は、数多くの専門家作業部会(Expert Working Groups: EWGs / Implementation Working Groups: IWGs)に参加しています。これらの作業部会では、科学的・技術的な専門知識に基づき、以下のような活動を行っています。
- 新規ガイドライン案の作成、既存ガイドラインの改訂に関する議論への参加
- ガイドラインに関するQ&Aや補遺文書、研修資料などの作成
- 日本の医薬品開発・審査の経験や科学的データ、規制状況を議論に提供し、国際的に受け入れられ、かつ実効性のあるガイドライン策定に貢献
3. 国内への情報提供と導入:
ICHで合意・採択されたガイドラインは、原則として各規制当局が国内の規制体系に導入することになっています。PMDAは、厚生労働省と連携し、ICHガイドラインの内容を反映した国内通知の発出などを通じて、国内での適切な実施を推進しています。
また、PMDAは、国内の製薬企業や研究機関などの関係者に対し、ICHの最新動向やガイドラインの内容、その背景などについて、説明会や研修などを通じて情報提供を行い、理解促進を図っています。
4. 情報発信拠点としての役割:
PMDAの公式ウェブサイトは、日本におけるICH情報の重要なハブとなっています。ウェブサイトでは、以下のような情報が網羅的に公開されており、誰でもアクセス可能です。
- ICHの組織、活動概要、会議に関する情報
- 全てのICHガイドライン(原文:英語)
- 主要なガイドラインの公式な日本語訳
- ガイドラインに関するQ&A(原文・和訳)
- ICHに関連する国内通知
- ICHに関する研修資材や報告書
このように、PMDAはICHの意思決定からガイドラインの策定、国内実施、情報提供に至るまで、多岐にわたる活動を通じて、医薬品規制の国際調和を推進し、日本の医薬品開発力の強化と、より質の高い医薬品を迅速に患者さんへ届けることに貢献しています。
参照
https://www.pmda.go.jp/int-activities/int-harmony/ich/0014.html
