臨床試験の一般指針
Table of Contents
1. 本指針の目的
医薬品開発戦略と薬効評価方法の発展に伴い、ICH(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use)に加盟している日・米・ EU 三極各々において臨床試験及び臨床開発方法の手順に関する一般指針が制定されてきた。本指針は他の ICH ガイドラインのみならず、これら三極の一般指針を基礎にして作成された。
本指針の目的は次のとおりである。
- 新医薬品の個々の臨床試験及び包括的な開発戦略に関する国際的に受け入れられる原則と具体的なあり方を記述すること。
- 臨床試験に関する一般的な原則、進め方及び関連用語の定義について三極が共通の理解をすることにより、外国の臨床試験データの評価と受け入れを促すこと。
- 臨床試験に関連する ICH ガイドラインの概観を示し、利用者が目的に応じて適切なガイドラインを利用しやすくすること。関連する各ガイドラインを別表に示す。
- 臨床試験に関連する ICH ガイドラインで使用されている臨床試験に関係する用語の解説集(注)を別冊として提供し、どのガイドラインでどの用語が使われているかを示すこと。
記載を簡潔にするために、本指針では「医薬品(場合により薬物)」を"drug"に対応する一般的な用語として使用しており、特に使い分ける必要がある場合を除き、"investigational (medicinal) product"(治験薬、場合により被験薬)、"pharmaceutical(product)"と同義である。また、この用語はワクチンや他の生物学的製剤も含むものである。本指針に示された原則は、他の臨床研究(例:放射線療法、精神療法、手術、医療機器、代替療法)にも適用しうる。
2. 一般的原則
2.1 被験者の保護
治験における被験者の安全確保に関する原則と具体的なあり方は、「医薬品の臨床試験の実施の基準(平成 9 年厚生省令第 28 号)」及び関連する通知(平成 9 年 3 月 27 日薬発第 430 号薬務局長通知、平成 9 年 5 月 29 日薬審第 445 号等) (以下「新 GCP ガイドライン」という。)に述べられている。これらの原則はヘルシンキ宣言を起源としており、人を対象とする全ての医薬品の試験を実施するにあたって遵守されなければならない。
いかなる臨床試験も、その開始にあたっては、非臨床試験又は先行する臨床試験の結果から、予定されている臨床試験においてその治験薬が十分安全であることが示されていなければならない。所定の試験期間の臨床試験を開始するために計画されるべき動物での薬理学的及び毒性学的試験の目的と実施期間については ICH M3 ガイドライン(別表)で論じられている。バイオテクノロジー応用医薬品に対するそのような試験については ICH S6ガイドライン(別表)に記載されている。
医薬品開発の全期間を通じて、新たに得られる動物での毒性試験データ及び臨床試験データについては、十分な適格性を有する専門家により、常に被験者の安全との関わり合いの観点から検討、評価されなければならない。得られる所見によっては、被験者の安全を確保するため、将来の試験、又は必要であれば実施中の試験についても、適切な時期に変更を加える必要がある。
治験責任(分担)医師と治験依頼者は、治験審査委員会と共に、被験者の保護に対する責任を有する。これら当事者の具体的な責任は、ICH E6 ガイドライン(別表)に述べられている。
2.2 科学的な臨床試験のデザインと解析
臨床試験は、その目的を達成するために、適切な科学的原則に従ってデザインされ、実施され、解析されるべきである。さらに、その試験結果は試験終了後適切に報告されなければならない。合理的な医薬品開発の本質は、重要な問題を提起し、適切な試験によってその問題に答えることである。いずれの試験においても主要な目的は明確でなければならず、予めはっきりと記述されていなければならない。
臨床試験は、その試験の臨床開発期間における実施時期及び目的によって表 1 のとおりに分類可能である。(ただし表 1 は治験の種類を網羅的に示したものではない。)先行する試験の成果が以降の試験の計画に当然影響を与えるはずであるという基本的な考え方があるからこそ、医薬品の臨床試験は段階的に進められるのである。新たに得られたデータによって、しばしば開発戦略の修正が必要になることもある。例えば、検証的試験の結果により臨床薬理試験の追加実施を行う必要性が示唆されることがある。
また、ICH E5 及び E6 ガイドライン(別表)に従って試験が実施されていれば、そのような外国の臨床試験データを入手、活用することにより、他の ICH 加盟国で同様なデータを繰り返し収集することが必要なくなるであろう。(ICH E5 ガイドライン(別表)参照)。
表1.目的による臨床試験の分類
| 試験の種類 | 試験の目的 | 試験の例 |
| 臨床薬理試験 |
|
|
| 探索的試験 |
|
|
| 検証的試験 |
|
|
| 治療的使用 |
|
|
3. 開発の方法
この章においては、開発計画及びそれを構成する個々の試験に関する論点及び考察について説明する。
3.1 開発計画に関して考慮すべき点
3.1.1 非臨床試験
非臨床試験について、その内容及び臨床試験との関係からの実施時期を決定する際に考慮すべき点として次のようなものが挙げられる。
- 個々の患者に対する投与期間及び総投与量
- 医薬品の特徴(例:長い半減期、バイオテクノロジー応用医薬品)
- 治療対象とする疾患又は症状
- 特別な集団における使用(例:妊娠可能な女性)
- 投与経路
臨床試験の実施を支持するために毒性、薬理及び薬物動態を含めた非臨床試験からの情報が求められていることについては、ICH M3 及び S6 ガイドライン(別表)において言及されている。
3.1.1.1 安全性試験
人での最初の試験における投与量は、臨床試験への移行前に実施されるべき非臨床試験での薬物動態、薬理学的及び毒性学的評価を注意深く考察した上で決定されなければならない(ICH M3 ガイドライン(別表))。
初期の非臨床試験は、人に対する初回投与量及び安全な投与期間を選択するために十分な情報、及び新医薬品の生理学的及び毒性学的作用についての情報が得られるものでなければならない。
3.1.1.2 薬理及び薬物動態試験
初期段階での臨床開発の根拠と方向性は、非臨床試験での薬物動態学的及び薬理学的プロフィール、即ち以下の情報に基づいて決定される。
- 主要な薬効の薬理学的根拠(作用機序)
- 用量―反応又は濃度―反応関係と作用持続時間
- 可能性のある臨床投与経路に関する試験
- 主要な臓器における薬理学的作用及び生理学的反応を含む全身的な一般薬理作用
- 吸収、分布、代謝及び排泄に関する試験
3.1.2 治験薬の品質
臨床試験で使用される製剤については、可能な限りバイオアベイラビリティについての情報も含め、その特性が十分に明らかにされていなければならない。製剤は治験薬の開発段階に応じた適切なものでなければならない。理想的には、用量幅を検討する一連の試験の実施に必要な十分な量の製剤が供給されることが望ましい。医薬品の開発期間中は、異なった種類の製剤が試験されることもある。生物学的同等性試験又は他の方法により確認された各製剤間の関連性は、開発計画全体にわたる臨床試験成績を解釈する際に重要である。
3.1.3 臨床開発における相
「医薬品の臨床開発は四つの逐次的な相(第Ⅰ相-第Ⅳ相)から成り立つ」と言われることがある。しかし、ある種の臨床試験は複数の相において実施されることもあることから、開発の相という概念が臨床試験の分類の基礎としてふさわしくないことを認識するのは重要である(図1参照)。2.2 に記した試験の目的による分類がより望ましい。相という概念は一種の記述表現であり、要求されていることそのものではないことを認識することが重要である。また、医薬品によっては典型的な開発順序が不適切、又は不必要であったりすることから、逐次的な相とは試験が決まった順序で行われることを意味しているわけではないことを認識することも重要である。例えば、臨床薬理試験は一般的に第Ⅰ相で行われるが、他の三つの相で実施されることも多い。しかし時にはそのような試験も第Ⅰ相試験と称される。図1は、試験の種類と開発の相という二つの分類方法の間の、密接ではあるが必ずしも一致しない関係を示している。図 1 の丸印(白丸、黒丸)の分布から、試験の種類が開発の相と同義ではないことが分かる。
図1
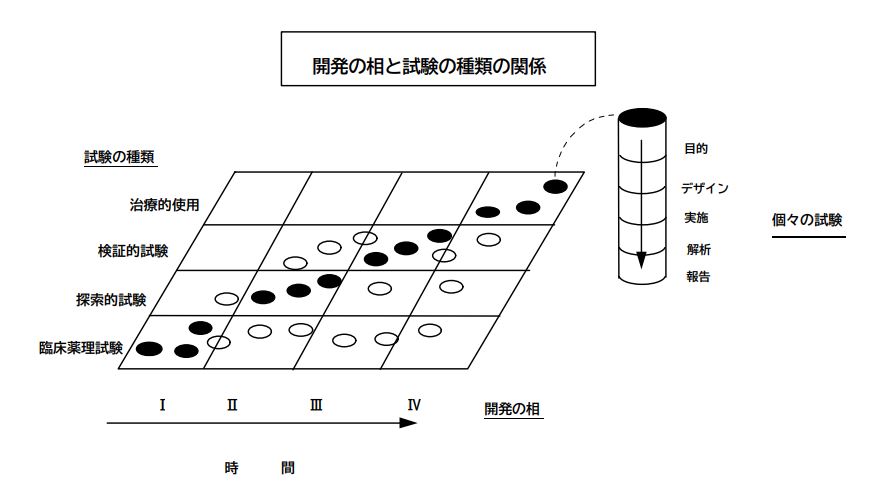
図1 この図は、開発の相と、ある医薬品の臨床開発に際し実施される目的別試験の種類との関係を表す。黒丸はある開発の相で最も一般的に実施される試験を示し、白丸はその相で実施されることが比較的まれな試験を示す。それぞれの丸は個々の試験を表し、右側のカラムはそれぞれの試験の構成要素とその順序を表す。
理想的には、医薬品開発は論理的で段階的な手続きにより進められる。その過程においては、小規模な初期の試験から得られた情報が、より後期の、より大規模な結論付けのための試験の計画及び根拠づけに用いられる。新医薬品を効率的に開発するためには、開発の初期の段階で治験薬の重要な特徴を見極め、それに基づいて適切な開発計画を立案することが必要不可欠である。
ごく初期の試験では、短期的な安全性と忍容性の初期評価が得られ、また、初期の治療効果の探索的試験に適切な用法・用量を選択するのに必要な薬力学及び薬物動態学上の情報が得られる。より後で実施される検証的試験は、一般的に、より大規模でより長期の試験であり、より多様な患者の集団を対象とする。用量-反応関係の情報は、開発の全ての段階で、すなわち初期の忍容性試験、短期の薬力学試験及び大規模な有効性を確認するための試験から得られるべきものである(ICH E4 ガイドライン(別表)参照)。開発期間を通じて、新たなデータが得られた結果、通常であればより前の相で行われるべき試験を追加する必要性が生じることがある。例えば、より後の試験で得られた血中濃度データが薬物間相互作用の試験の必要性を示唆したり、あるいは有害作用が追加の用量探索試験や非臨床試験の実施の必要性を示唆することがある。さらに、既承認医薬品について新たな承認(例:新効能)申請のための開発を行う場合には、薬物動態試験又は探索的試験は、開発過程の第Ⅰ相又は第Ⅱ相で実施されるものと考えられる。
3.1.3.1 第Ⅰ相(最も代表的な試験:臨床薬理試験)
第Ⅰ相は、治験薬を初めて人に投与することから開始される。
通常、臨床薬理試験は、第Ⅰ相において実施されると見なされるが、開発の他の時点で行われることもある。開発におけるこの相の試験は、通常、治療効果をみることを目的としない試験であり、健康な志願者又は特定のタイプの患者、例えば、軽度の高血圧症の患者で実施される。強い毒性を持った医薬品、例えば抗悪性腫瘍薬では、通常、患者を対象として試験が行われる。この相の試験では、対照を置かなかったり、治験薬投与前値との比較を行ったり、又は観察の信頼性を向上させるために無作為化及び盲検化を行ったりすることもある。
第Ⅰ相において実施される試験は、通常、次の一つ又はその組合せの観点から行われる。
a) 初期の安全性及び忍容性の推測
治験薬を初めて人に投与する試験は、通常、後の臨床試験のために必要と想定される用量範囲の忍容性を決定し、予期される副作用の性質を判断するために行われる。これらの試験には、通常、単回及び反復投与が含まれる。
b) 薬物動態
薬物の吸収、分布、代謝、排泄に関する特徴の検出は、開発計画全体を通して行われる。
これらの特徴を予備的に見出すことは、第Ⅰ相において実施される試験の重要な目的の一つである。薬物動態は独立した試験において評価されることもあれば、有効性・安全性・忍容性の試験の一部において評価されることもある。薬物動態試験では、薬物のクリアランスを評価し、未変化体又は代謝物の蓄積の可能性及び薬物相互作用の可能性を予期することが特に重要である。また、より特殊な問題に答えるために、後の相でいくつかの薬物動態試験が行われることもよくある。経口投与される医薬品の多く、特に薬物放出が制御されるべく設計された製剤においては、バイオアベイラビリティに対する食事の影響を検討する試験は重要である。代謝・排泄障害(腎・肝疾患)を有する患者、高齢者、小児、女性及び人種のサブグループのような部分集団における薬物動態の情報を得ることも考慮しなければならない。多くの医薬品については、薬物間相互作用の試験は重要である。これらは一般的に第Ⅰ相より後の相で実施されるが、代謝と相互作用の可能性を検討する動物試験及び in vitro 試験の結果によっては、より早期に実施されることもあり得る。
c) 薬力学的な評価
医薬品及びエンドポイントによっては、薬力学試験及び薬物の血中濃度と反応に関する試験(薬物動態/薬力学的試験)を、健康な志願者又は目標とする疾患を有する患者を対象として行うことがある。患者を対象とする試験で、適切な指標がある場合には、薬力学的データから薬効及び予想される有効性の初期の推測が可能である。これらの薬力学的データは後の試験における用法・用量の設定に役立つことがある。
d) 初期の薬効評価
薬効又は見込まれる治療上の利益の予備的検討が、副次的な目的として第Ⅰ相で行われることがある。一般的にはこのような試験はより後の相で行われるが、この初期的段階に患者に短期間治験薬を投与することにより薬効が容易に測れる場合には、この相での実施が適切なこともあろう。
3.1.3.2 第Ⅱ相(最も代表的な試験:探索的試験)
第Ⅱ相は、通常、患者における治療効果の探索を主要な目的とする試験を開始する段階である。
初期の探索的臨床試験では、同時対照や投与前の状態(ベースライン)との比較等、様々な試験デザインが用いられる。それに続く試験として、特定の適応に対するその治験薬の有効性と安全性を評価するために、通常、無作為化同時対照比較試験が実施される。第Ⅱ相における試験は、比較的均質な集団になるように比較的狭い基準に従って選択された患者を対象として注意深く観察しながら行われるのが普通である。
第Ⅱ相の重要な目的は、第Ⅲ相で行われる試験の用法・用量を決定することである。第Ⅱ相の初期的試験では、用量反応の初期的推測のために、用量の漸増デザイン(ICH E4ガイドライン(別表)参照)がしばしば用いられる。それに続く試験では、並行用量反応デザイン(第Ⅲ相で実施されることもある。)を用いて目的とする適応に対する用量-反応関係が確認されることになる。検証的な用量反応試験は、第Ⅱ相で実施されることもあれば第Ⅲ相で実施されることもある。第Ⅱ相での用量は、通常、第Ⅰ相の最高用量より低用量であるが、常にそうであるとは限らない。
第Ⅱ相で実施される試験のその他の目的としては、その後に実施する第Ⅱ相や第Ⅲ相試験において用いられる見込みのあるエンドポイント 、治療方法(併用療法を含む。)、対象となる患者群(例:軽症例か重症例か)を評価することが挙げられる。これらの目的はデータを部分的に吟味する探索的解析や、試験に複数のエンドポイントを設定すること等により達成されるであろう。
3.1.3.3 第Ⅲ相(最も代表的な試験:検証的試験)
第Ⅲ相は、通常、治療上の利益を証明又は確認することを主要な目的とする試験を開始する段階である。
第Ⅲ相に実施される試験は、意図した適応及び対象患者群においてその治験薬が安全で有効であるという第Ⅱ相で蓄積された予備的な証拠を検証するためにデザインされる。このような試験は、承認のための適切な根拠となるデータを得ることを意図している。第Ⅲ相では、用量―反応関係をさらに探索する試験、より広い対象患者や病態の異なるステージでの医薬品の使用又は他剤との併用を検討する試験を実施することもある。長期投与を意図した医薬品については、投与期間を延長した試験は、第Ⅱ相から開始することもあるが、通常は第Ⅲ相で実施される(ICH E1 ガイドライン(別表)参照)。ICH E1 及び E7ガイドライン(別表)では、長期投与される医薬品及び高齢者に用いられる医薬品についての臨床上の安全性データベースに係る考慮点について記述している。第Ⅲ相で実施されるこれらの試験において、医薬品の適切な使用法を支持するのに必要な情報(正式な製品情報)を得ることになる。
3.1.3.4 第Ⅳ相(多様な試験:治療的使用)
第Ⅳ相に実施される試験は、医薬品の承認後に始まる。それ以前に医薬品の安全性、有効性が示され、用量が設定されてはいるが、治療的使用での試験はさらにそれ以上の知見を得るためのものである。
第Ⅳ相での試験は、医薬品承認後に行われるすべての試験(ルーチンの市販後調査を除く。)であり、承認された適応に関連したものである。これらの試験は、必ずしも承認には必要でないと考えられるが、その医薬品の最適な使用法を明らかにする上で重要である。
第Ⅳ相での試験は、様々な形態をとるかもしれないが、適切な科学的な目的を有していなければならない。一般的に行われる試験には、追加的な薬物相互作用試験、用量―反応試験、又は安全性試験、そして承認された適応疾患における使用を支持するための試験(例:死亡率/罹病率に係る試験、疫学試験)が含まれる。
3.1.3.5 新たな用法等を目指す開発
最初の承認後も、新効能若しくは効能の変更、新用法・用量、新投与経路、又は追加の患者集団での試験を行うことにより開発が続けられる場合もある。新用量、新剤型、新配合を検討する場合には、新たな開発計画のもと、追加の臨床薬理試験が必要となることがある。
当初の開発計画又は治療的使用から得られたデータを利用することにより、ある種の試験を省略できることがある。
3.1.4 特別に考慮すべき点
開発計画の一部に特殊な状況や特別な集団が含まれている場合は、それらについて検討を要する場合が多い。
3.1.4.1 薬物代謝に関する試験
主要な活性代謝物については、これを同定し、その詳細な薬物動態試験を実施しなければならない。開発計画の中で代謝に関する評価試験を行う時期は、各々の薬物の性質により決まる。
3.1.4.2 薬物間相互作用
代謝のプロフィール、非臨床試験の結果や類似薬物についての情報から薬物間相互作用が示唆される場合には、薬物間相互作用に関する検討を実施することが特に勧められる。高い頻度で併用される医薬品については、非臨床試験及びもし適切であれば臨床試験で薬物間相互作用試験を行うことも通常は重要である。他の薬物の吸収や代謝を変える(ICH E7ガイドライン(別表)参照)ことが知られている医薬品や他の薬物の作用により代謝や排泄が変化する医薬品では、特に重要である。
3.1.4.3 特別な集団
一般的な集団の中のいくつかのグループについては、開発中に特殊なリスク・ベネフィットを考慮する必要があるという理由から、また、一般の成人と比較して投与量又は投与スケジュールを変更する必要があるという理由から、特別な試験が必要な場合がある。腎障害及び肝障害の患者に対して薬物動態学的検討を行うことは、変化しているかもしれない代謝又は排泄の影響を評価するために重要である。他の ICH ガイドラインでは、高齢者(E7 ガイドライン(別表))、及び異なる人種(E5 ガイドライン(別表))についてこのような問題を論じている。特別な集団を対象とした臨床試験の妥当性を支持するための非臨床安全性試験の必要性については ICH M3 ガイドライン(別表)に記載されている。
社会的弱者のインフォームドコンセント及び慎重さが求められるその後の手続きに関する倫理的な考慮点には特に注意を払うべきである(ICH E6 ガイドライン(別表)参照)。
a)妊婦における検討
一般に、妊婦は、妊娠時の使用を目的としていない医薬品の治験からは除外されるべきである。治験薬の投与中に被験者が妊娠した場合には、投与を問題なく中止できるのであれば、治験薬の投与を中止すべきである。この場合には、妊娠、胎児、出生児の追跡評価を行うことは重要である。同様に、妊娠中に使用される医薬品につき妊婦が参加する臨床試験でも、妊娠、胎児、出生児の追跡評価が非常に重要である。
b)授乳婦における検討
薬物又はその代謝物の乳汁への排泄については、必要に応じて検討すべきである。授乳婦が試験に加わった際は、授乳されている乳児に対する薬物の影響についても観察すべきである。
c)小児における検討
ある医薬品に関するその時点での知見、及び成人や他の年齢の小児におけるデータの外挿の可能性により、求められる試験の程度・範囲が決まる。医薬品によっては開発の初期段階から小児に使用されることもある。(ICH M3 ガイドライン(別表)参照)。
小児に使用されることを目的とした医薬品の試験では、適切な年齢の集団を対象として検討を行うべきである。臨床開発が小児を対象とする試験を含む場合には、通常、なるべく年長児から開始し、ついで年少児、幼児と試験を拡大するのが適切な方法である(ICH E6ガイドライン(別表)参照)。
3.2 個々の臨床試験において考慮すべき点
臨床試験の目的、デザイン、実施、解析、報告を立案する際には、以下の重要な原則に従って行われるべきである(別表の ICH ガイドライン参照)。目的から報告までの各項目は、試験開始前に治験実施計画書に明確に規定されなければならない(ICH E6 ガイドライン(別表)参照)。
3.2.1 目的
試験の目的は明確に記述されていなければならない。目的においては、安全性及び(又は)有効性及び(又は)薬力学的パラメータの評価及び薬理学的、生理学的、生化学的効果の評価について、それが探索的なものなのか、検証的なものなのかが記載されることがある。
3.2.2 デザイン
目的とする情報を得るために適切な試験デザインを選択しなければならない。試験デザインとしては、並行群間試験、クロスオーバー試験、要因試験、漸増法試験、固定用量用量反応試験などがある(ICH E4、E6、E9、E10 各ガイドライン(別表)参照)。試験目的を達成するために、適切な対照の使用と十分な数の被験者が必要である。主要及び副次的エンドポイントとその解析法は明確に記述されていなければならない(ICH E9 ガイドライン(別表)参照)。臨床所見、症状、臨床検査値上の変動によって有害事象をモニターする方法も記載されなければならない(ICH E3 ガイドライン(別表)参照)。また、治療を中止した患者のフォローアップの手順は治験実施計画書に明記されなければならない。
3.2.2.1 被験者の選択
被験者集団の選択(例:開発初期において健康志願者を対象とするか、癌患者又は他の特別な被験者を選択するか)にあたっては、先行する非臨床試験及び臨床試験の知見とともに、開発の段階や検討される適応症も考慮されるべきである。
初期段階の試験で被験者となる患者集団又は健康志願者集団は、厳格な選択条件によって狭い範囲に限定されるだろうが、医薬品の開発段階が進むにつれて、被験者の集団はその医薬品が目標とする患者集団を反映するよう拡大されるべきである。
開発の段階及び安全性に対する懸念の程度によっては、厳密に監視された環境(例:入院下)において試験を行うことも必要であろう。一般的な原則として、被験者は同時に一つ以上の臨床試験に参加すべきではないが、例外が正当化される場合もある。安全の確保及び持ち越し効果の排除のための十分な休薬期間を置かずに被験者が臨床試験に繰り返し組み込まれることがあってはならない。一般に、妊娠可能な女性は、試験に参加する際には十分に効果的な方法で避妊すべきである(ICH M3 ガイドライン(別表)参照)。男性被験者に関しては、医薬品の投与がその性的伴侶や子孫に対し及ぼしうる危険について考慮されるべきである。そのような可能性が示唆される場合(例:変異原性や生殖毒性の可能性のある医薬品を用いる試験)は、適切な避妊法に関する規定が試験に含まれるべきである。
3.2.2.2 対照群の選択
試験には適切な対照群が必要である。比較にはプラセボ、無処置、実薬対照又は被験薬の異なった用量等が用いられる。対照の選択は、何よりも試験の目的によって決められる(ICH E9、E10 各ガイドライン(別表)参照)。ヒストリカル(外部)対照を用いることが正当化できる場合もあるが、誤った推論の可能性を最小限にすることに特に注意が必要である。
3.2.2.3 被験者数
試験の規模は、検討対象となる疾患、試験の目的及び試験のエンドポイントに左右される。被験者数に関する統計学的な判断は、期待される治療効果の大きさ、データのバラツキ、許容できる一定の危険率(ICH E9 ガイドライン(別表)参照)、必要な患者サブセット、及び副次的エンドポイントに関する情報をどの程度必要とするのかに基づいて行われるべきである。状況によっては、薬剤の安全性を確立するためにより大きなデータベースが必要になることもある。 ICH E1 及び E7 ガイドライン(別表)では、新しい適応について、安全性を評価するためのデータベースに最低限必要な症例数を記載している。しかし、これらの数は絶対的なものと考えるべきではなく、また、時には不十分なこともある(例:健康者に長期間にわたって使用されることが見込まれる場合)。
3.2.2.4 反応変数
反応変数は、試験開始前に規定され、観察及び定量化の方法を具体的に示すものでなければならない。可能かつ適切である限り、客観的な観察方法が用いられるべきである(ICH E9 ガイドライン(別表)参照)。
試験のエンドポイントは、薬物動態パラメーター、薬力学的評価指標、有効性及び安全性に関連する医薬品の効果を評価するために選択された反応変数である。主要なエンドポイントは臨床上意味のある効果を反映すべきであり、通常、試験の主要な目的に基づいて選択される。副次的なエンドポイントは、医薬品のその他の効果を評価するための項目であり、主要なエンドポイントに関連していることもあれば関連していないこともある。エンドポイントとその解析方法は、治験実施計画書に予め定めておかなければならない。
代用エンドポイントは、臨床上重要な結果に関連づけることを意図したエンドポイントであるが、それ自体が臨床上のベネフィットを測るものではない。代用エンドポイントは、適切な場合(代用エンドポイントを使うことにより、十分合理的に臨床上の結果を予測しうる場合又は臨床上の結果を予測しうることがよく知られている場合)には、主要なエンドポイントとして用いることができる。
主観的なものであれ客観的なものであれ、エンドポイントの評価に用いられる方法は、バリデートされたものでなければならず、かつ正確性、精度、再現性、信頼性及び反応性(経時変化に対する感度)に係る適切な基準を満たすものでなければならない。
3.2.2.5 偏りを最小にする方法又は評価する方法
治験実施計画書には、治療群への割付方法及び盲検化の方法について明記しなればならない(ICH E9、E10 各ガイドライン(別表)参照)。
a) 無作為化
比較試験の実施において、無作為化は、試験群間の比較可能性を保証し、被験者選択時のバイアスの可能性を最小とする好ましい方法である。
b) 盲検化
盲検化は、試験結果に偏りを生じさせる危険性を減少又は最小化する重要な方法である。
プラセボや他の治療的介入の種類を識別不可能にする方法を用いることにより、割付けられた治療を被験者が知らされない試験を単盲検試験という。さらに、被験者に対する処置、臨床的評価及びデータ解析等に関わる治験責任(分担)医師及び治験依頼者のスタッフも割付けられた治療法を知らない試験を二重盲検試験という。
c) 服薬状況
治験実施計画書には被験者の治験薬服薬状況をどのように評価するかを明記しなければならず、また、実際の服薬状況を記録しなければならない。
3.2.3 実施
臨床試験は、本ガイドラインに記載してある原則、ICH E6 ガイドライン及び臨床試験に係る他の ICH ガイドライン(別表参照)に概説されている関連原則に従って実施されなければならない。治験実施計画書の遵守は必須である。治験実施計画書の変更を要する場合は、その変更の合理的理由が改訂治験実施計画書に明確に記載されなければならない(ICH E6 ガイドライン(別表)参照)。試験期間中は有害事象について適切なタイミングでの報告が必須であり、報告は記録されなければならない。規制当局に対する安全性データの迅速な報告、安全性情報の内容及びプライバシーの保護、データの機密性に関しては、他の ICH ガイドライン(ICH E2A、E2B 及び E6 ガイドライン(別表))を参照すること。
3.2.4 解析
治験実施計画書に記載される解析計画は、被験者の割付方法、反応変数の測定方法、検証すべき特定の仮説及び早期中止や治験実施計画書の遵守違反等の通常起こりうる問題に対する解析上の対処法を考慮した上で、試験の目的やデザインに適するものでなければならない。計画された中間解析を行うタイミングを含め、採用される統計学的方法を治験実施計画書に記載しておかなければならない(ICH E3、E6 及び E9 ガイドライン(別表)参照)。
臨床試験の結果は、治験実施計画書に予め記載された解析方法に従って解析され、試験計画からの逸脱例については全て総括報告書に記述されなければならない。試験計画の立案(ICH E6 ガイドライン)、解析計画、統計解析(ICH E9 ガイドライン)及び総括報告書(ICH E3 ガイドライン)については各ガイドライン(別表)の詳細な解説を参照すること。
通常、臨床試験は完了することを前提に実施されるが、早期に中止する可能性が正式に認められている試験も一部にある。このように早期中止を認める試験の場合には、全体的な統計学的有意性について、及び治療効果の大きさに関する推定値を調整する必要性について統計学的に十分に配慮しつつ、その旨を治験実施計画書に明記する必要がある(ICH E9ガイドライン(別表)参照)。
安全性データはすべての臨床試験において収集され、一覧表にまとめられなければならない。集積された有害事象は重篤度と因果関係に従って分類されなければならない(ICH E2A ガイドライン(別表)参照)。
3.2.5 報告
臨床試験の総括報告書は、他の ICH ガイドライン(ICH E3、E6 ガイドライン(別表)参照)に記載されている方法に従って適切に作成されなければならない。
参照
https://www.pmda.go.jp/int-activities/int-harmony/ich/0030.html
https://note.com/realworld/n/ne5b1e3ddfbd5
