新しいお薬が私たちの手元に届くまでには、そのお薬が本当に安全で、ちゃんと効くのかどうかを、国の機関(規制当局)が厳しくチェックする「承認審査」というステップがあります。この審査のために、製薬会社はたくさんの資料を提出する必要があります。
昔は、国ごとに提出する書類の形式がバラバラで、同じお薬を複数の国で承認してもらおうとすると、国ごとに書類を作り直さなければならず、とても大変でした。時間もお金もかかり、新しいお薬を早く届けたいのに、書類作成がボトルネックになってしまうことも…。
そこで登場したのが、「コモン・テクニカル・ドキュメント(CTD:Common Technical Document)」です!
Table of Contents
CTDって、いったい何?
CTDは、お薬の承認申請に必要な書類を、世界共通の構成(フォーマット)でまとめましょうという国際的なルールのことです。日本、アメリカ、ヨーロッパなど、多くの国がこのルールを採用しています。
この共通ルールを決めたのが**ICH(日米EU医薬品規制調和国際会議)**という、医薬品規制に関する国際的な話し合いの場です。ICHのおかげで、お薬の品質や安全性、有効性を評価するための技術的な基準はかなり統一されてきましたが、申請書類の「形」までは揃っていませんでした。CTDは、その最後のピースを埋めるものなのです。
CTDのメリット
CTDを使うと、こんないいことがあります。
- 書類作成が楽になる!:一度CTD形式で書類を作れば、基本的な部分はどの国にも提出できます。書類作成にかかる時間と労力を大幅に減らせます。
- 電子申請 (eCTD) がしやすくなる!:データでの提出(電子申請)をスムーズに行えるようになります。
- 審査がスピードアップ!:審査する側(規制当局)も、構成が統一されているので内容を理解しやすく、審査が効率的に進みます。
- 国際協力が進む!:国境を越えて、お薬に関する情報のやり取りがしやすくなります。
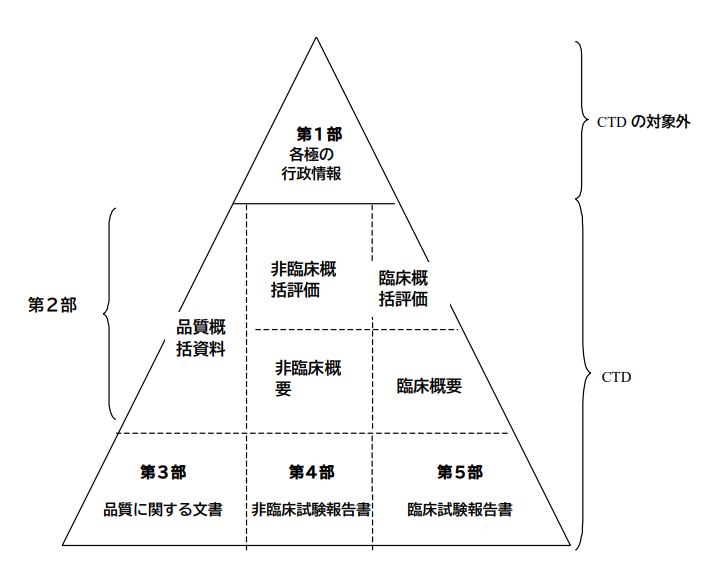
CTDのしくみ:情報は5つの「モジュール」に整理
CTDは、申請情報を大きく5つの「部(モジュール)」に分けて整理します。ちょうど、書類を整理する大きな棚に、テーマごとに引き出し(モジュール)があるイメージです。
- 第1部(モジュール1):各国固有の情報 🇯🇵🇺🇸🇪🇺
- ここには、申請先の国ごとに必要な書類(申請書そのものや、日本でいう「添付文書」の案など)を入れます。内容は国によって異なります。
- 第2部(モジュール2):申請内容のまとめ(サマリー) 📑
- ここがCTDの「顔」とも言える部分です。お薬がどんな種類で、どう作用するのか、どんな効果が期待されるのか、といった全体の概要を1ページ程度でまず説明します。
- さらに、お薬の品質、非臨床試験(動物などで行う試験)、臨床試験(ヒトで行う試験)の結果について、それぞれ専門的な要約(概括評価、概要文、概要表)をまとめて記載します。審査員がまず全体像を把握するための重要なパートです。
- 第3部(モジュール3):お薬の「品質」に関する詳しい情報 🔬🧪
- お薬そのものや、その製造方法、安定性など、「品質」に関する詳細なデータや報告書をまとめます。
- 第4部(モジュール4):非臨床試験(動物実験など)の詳しい報告書 🐁
- お薬の効き目や安全性を、動物実験などで調べた結果(非臨床試験)の詳細な報告書をまとめます。
- 第5部(モジュール5):臨床試験(ヒトでの試験)の詳しい報告書 🧑⚕️
- 実際にお薬をヒトに使ってみて、その有効性や安全性を確認した試験(臨床試験)の詳細な報告書や関連資料をまとめます。
ポイント: モジュール2から5までは、どの国に申請する場合でも共通の構成になります。これがCTDの最大の利点です!
ICHコモン・テクニカル・ドキュメントの概念図
書類作成のルール:読みやすく、分かりやすく!
CTDを作成するときには、いくつか守ってほしいルールがあります。
- 分かりやすさ第一!:専門的な内容ですが、審査員がスムーズに理解できるよう、あいまいな表現を避け、明確で分かりやすい記述を心がけましょう。
- 読みやすい形式で!:
- 用紙サイズはA4(日本、EU)またはレターサイズ(米国)どちらでも印刷できるよう、余白を適切にとります。特に左側の余白は、綴じても文字が隠れないように十分確保しましょう。
- 文字の大きさや種類(フォント)は、コピーしても読めるように配慮します(例:日本語ならMS明朝10.5ポイント程度)。
- 各モジュールの最初のページを「1ページ」とし、すべてのページに番号を振ります。
- 専門用語の略語を使う場合は、最初に必ず正式名称を示しましょう。
- 参考文献は、国際的なルール(バンクーバー宣言)に従って記載します。
どんなお薬の申請に使うの?
このCTDのルールは、主に**新しい有効成分を含む医薬品(バイオテクノロジーを使ったお薬も含む)**の承認申請で使われます。
注意点: CTDはあくまで「書類の構成」を決めるルールであり、「どんな試験をしなければならないか」を決めるものではありません。また、基本的な構成(モジュール1〜5の枠組み)は変えられませんが、モジュール2の「非臨床」や「臨床」の概要部分については、内容がより分かりやすくなるように、個々の資料の表現方法を工夫することは可能です。
まとめ:CTDで、より良い医療の未来へ
CTDは、国境を越えて新しいお薬の承認申請をスムーズにするための、画期的な共通言語です。これにより、製薬会社は開発に集中でき、規制当局は効率的に審査でき、最終的には患者さんが新しい治療法に早くアクセスできるようになることが期待されています。今後、電子化されたeCTDの活用も進み、さらに効率的な申請・審査が進んでいくでしょう。
付録1:ICH-M6ガイドライン
医薬品の承認申請のための国際共通化資料
コモン・テクニカル・ドキュメント(CTD)の構成
ガイドラインの目的
本ガイドラインは、承認申請のために規制当局に提出される適切に構成されたコモン・テクニカル・ドキュメント(CTD)の作成について、合意に達した共通の様式を示したものである。申請資料作成のための共通様式は、医薬品承認申請のための文書編集に要する時間及び資源を著しく軽減し、電子申請の準備を容易にするであろう。
また、規制当局による審査及び申請者とのコミュニケーションは、共通の構成要素から成る標準化された文書により促進されるであろう。さらに、規制当局間の規制情報の交換を容易にするであろう。
背景
ICH(日米EU医薬品規制調和国際会議)により、医薬品の承認申請のための技術的要件については、三地域間で相当の調和が達成されてきた。しかしながら、現在まで承認申請文書の構成に関する調和が達成されてきていなかった。各地域は、提出書類中の申請資料の配列並びにサマリー及び表作成のためのそれぞれの要件を有している。日本では、申請者は、承認申請資料をまとめた資料概要を作成しなければならない。欧州では、エキスパート・レポート(Expert Report)及び概要表(Tabulated Summary)が要求され、概要文(Written Summary)を提出することが推奨されている。米国FDAには、新薬承認申請の様式及び内容に関するガイダンスがある。地域間で異なった承認申請資料の作成・編集の必要性を避けるため、本ガイドラインでは、三地域のいずれにおいても受入れ可能なCTDのための様式を提示する。
本ガイドラインの適用範囲
本ガイドラインは、主に新有効成分含有医薬品(バイオテクノロジー応用医薬品を含む。)の承認申請において示されるべき資料の構成について述べたものである。
本ガイドラインは、どのような試験の実施が要求されるのかを示すことを意図したものではなく、単に得られたデータに用いられる適切な様式を示しているに過ぎない。申請者は、本ガイドラインに示されているCTD全体の構成を変更すべきではないが、非臨床及び臨床概要においては、試験成績の理解及び評価を容易にするために、必要に応じ、申請資料の内容が可能なかぎり最も適切な表記となるよう、個々の資料の様式を変更することは差し支えない。
一般原則
基礎的データの審査を容易にし、審査官が申請内容を速やかに把握できるよう、資料内容の表現の仕方はCTD全体を通じてあいまいさを排し、わかりやすいものとすること。
本文及び表は、その内容がA4サイズ(EU及び日本)並びに8.5×11インチ(米国)の紙のいずれにも印刷できるだけの余白をもって作成すること。左側の余白を十分広くとり、綴じても情報が隠れないようにすること。本文及び表のフォントサイズは、複写後も十分に読みやすい大きさのスタイルと大きさとすること。本文の説明の記述には、英文の場合はTimes New Romanの12ポイント(注:日本語の場合はMS明朝の10.5ポイントに相当。)を推奨すること。各モジュールの最初のページをページ1とし、全ページにページ番号を付けること。一つのモジュールが複数の巻からなる場合は、各々の巻はページ1から始めることができること。各々のモジュールの中で用いられる頭文字及び略語は最初に定義すべきであること。引用文献は、生物医学誌への投稿のために必要な定型様式に関する1979年バンクーバー宣言に従って引用されるべきであること。
CTDの構成
CTDは5つの部(モジュール)で構成されている。第1部(モジュール1)については各地域に特異的な部分である。第2部から第5部まで(モジュール2から5まで)は、全ての地域への申請において共通となるよう意図されている。本ガイドラインに従うことにより、これら4つの部(モジュール)は規制当局に対して受入れ可能な様式で提供されることとなる。
第1部(モジュール1) 申請書等行政情報及び添付文書に関する情報
この部(モジュール)には、例えば、当該地域における申請書又は添付文書(案)といった各地域に特異的な文書が含まれる。この部(モジュール)の内容及び様式については、当該規制当局が定めることができる。
第2部(モジュール2) CTDの概要(サマリー)
第2部(モジュール2)は、薬理学的分類、作用機序及び申請する効能又は効果等の当該医薬品の全般的な概略から始めること。原則として、この緒言は1ページ以内にまとめること。
第2部(モジュール2)は、品質に関する概括資料、非臨床及び臨床に関する概括評価で構成すること。それに引き続き、非臨床試験に関する概要文及び概要表、並びに臨床概要を提出すること。これら概要の個々の構成については、CTD-品質に関する文書の作成要領に関するガイドライン(M4Q)、CTD-非臨床に関する文書の作成要領に関するガイドライン(M4S)、及びCTD-臨床に関する文書の作成要領に関するガイドライン(M4E)のそれぞれのガイドライン中に規定するものである。
第3部(モジュール3) 品質に関する文書
品質に関する資料を、CTD-品質に関する文書の作成要領に関するガイドライン(M4Q)に記載された様式で添付すること。
第4部(モジュール4) 非臨床試験報告書
非臨床試験報告書を、CTD-非臨床に関する文書の作成要領に関するガイドライン(M4S)に記載された順序で添付すること。
第5部(モジュール5) 臨床試験報告書
臨床試験報告書及び関連資料を、CTD-臨床に関する文書の作成要領に関するガイドライン(M4E)に記載された順序で添付すること。
付録2:CTDの各モジュールに含まれる情報
以下は、CTDの各モジュールに含まれる情報の詳細なリストです。
第1部(モジュール1):申請書等行政情報及び添付文書に関する情報
- 1.1 第1部(モジュール1)の目次
- 1.2 各地域に特異的な文書(例:申請書、添付文書(案))
第2部(モジュール2):CTDの概要(サマリー)
- 2.1 CTD全体の目次
- 2.2 緒言(医薬品の概要、1ページ以内)
- 2.3 品質に関する概括資料
- 2.4 非臨床に関する概括評価
- 2.5 臨床に関する概括評価
- 2.6 非臨床概要
- 薬理(概要文、概要表)
- 薬物動態(概要文、概要表)
- 毒性(概要文、概要表)
- 2.7 臨床概要
- 生物薬剤学及び関連する分析法の概要
- 臨床薬理の概要
- 臨床的有効性の概要
- 臨床的安全性の概要
- 個々の試験のまとめ
第3部(モジュール3):品質に関する文書
- 3.1 目次
- 3.2 データ又は報告書
- 3.3 参考文献
第4部(モジュール4):非臨床試験報告書
- 4.1 目次
- 4.2 試験報告書
- 4.3 参考文献
第5部(モジュール5):臨床試験報告書
- 5.1 臨床試験報告書及び関連情報の目次
- 5.2 臨床試験一覧表
- 5.3 臨床試験報告書及び関連情報
- 5.4 参考文献
